Bonjour,
J'ai un problème avec le paquet MOFA à la R.3.6. Veuillez voir ci-joint.

J'ai suivi les instructions d'installation mais cela ne change rien à la situation. Pourriez-vous m'aider à résoudre le problème.
Merci beaucoup de votre aide!
zxue
Bonjour,
J'ai un problème avec le paquet MOFA à la R.3.6. Veuillez voir ci-joint.
J'ai suivi les instructions d'installation mais cela ne change rien à la situation. Pourriez-vous m'aider à résoudre le problème.
Merci beaucoup de votre aide!
zxue
Bonjour,
L'installation de MOFA est en cours sur l'env R 3.6:
A priori ça devrait permettre de redescendre les dépendances Python manquantes dans l'environnement R.
cela ne change pas l'erreur de mon côté...
Ok merci,
Est-ce que vous pouvez fournir un petit script R qui permettrai de tester sur nos env de dev et preprod ?
(Genre le script minimal pour reproduire l'erreur facilement)
Bonsoir,
Deuxième essai:
Bonjour Zeyun,
Il semble exister deux version de MOFA v1 et v2:
On bascule sur la "v2" ?
Bonne journée
Bonjour Francois et David,
Thanks for your imput.
Here's the script R I run with cll_data. I haven't switch to MOFAv2 yet. Please see as follows,I put sessioninfo() incase it helps.
library(MOFA)
library(MOFAdata)
data("CLL_data")MOFAobject <- createMOFAobject(CLL_data)
Creating MOFA object from a list of matrices
please make sure that samples are stored in the columns and features in the rows...
lapply(CLL_data, dim)
$Drugs
[1] 310 200
$Methylation
[1] 4248 200
$mRNA
[1] 5000 200
$Mutations
[1] 69 200
data("CLL_covariates")
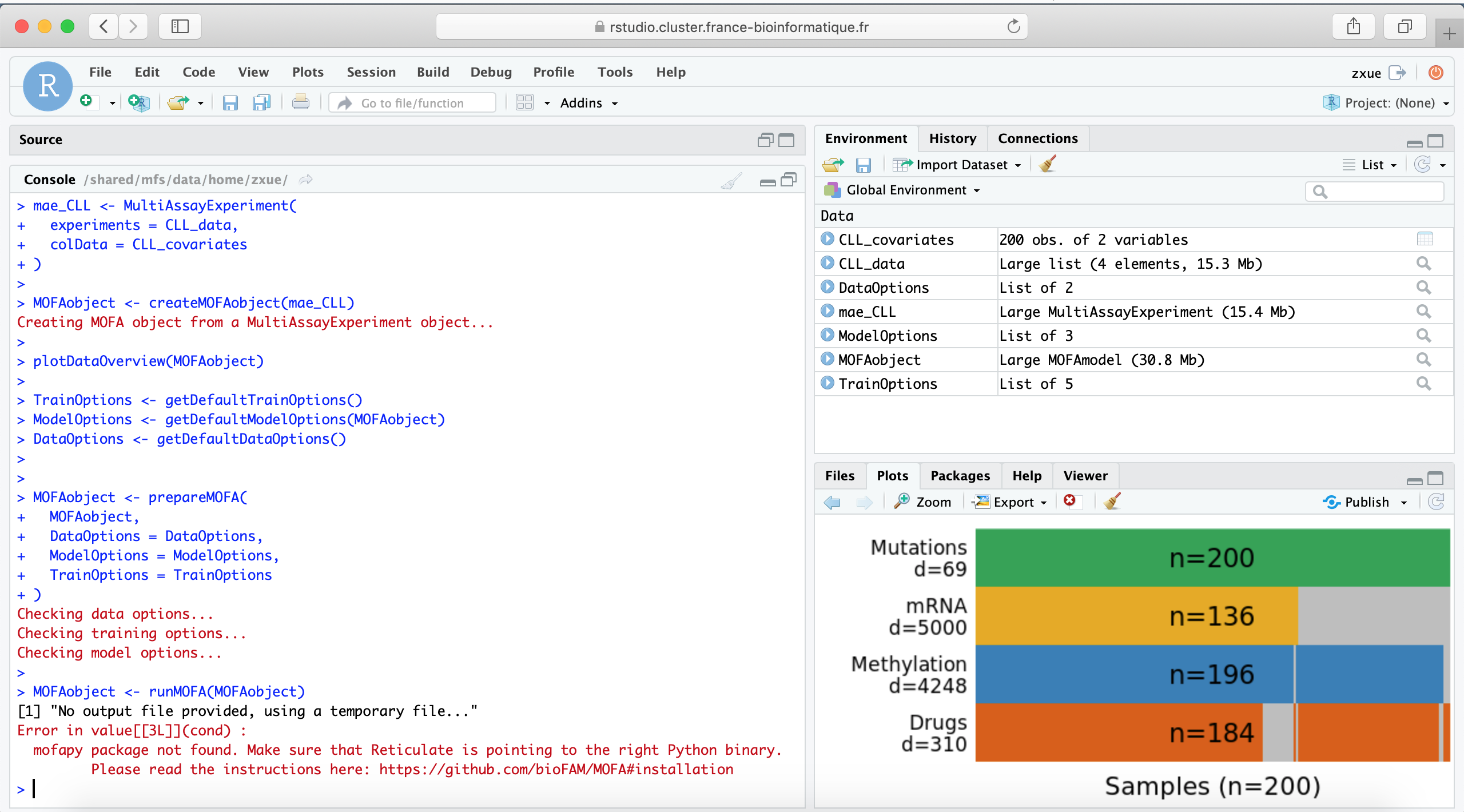
mae_CLL <- MultiAssayExperiment(
experiments = CLL_data,
colData = CLL_covariates
)
MOFAobject <- createMOFAobject(mae_CLL)
Creating MOFA object from a MultiAssayExperiment object...
plotDataOverview(MOFAobject)
DataOptions <- getDefaultDataOptions()
DataOptions
$scaleViews
[1] FALSE
$removeIncompleteSamples
[1] FALSE
ModelOptions <- getDefaultModelOptions(MOFAobject)
ModelOptions$numFactors <- 25
MOFAobject <- prepareMOFA(
MOFAobject,
DataOptions = DataOptions,
ModelOptions = ModelOptions,
TrainOptions = TrainOptions
)
Checking data options...
Checking training options...
Checking model options...
MOFAobject <- runMOFA(MOFAobject)
[1] "No output file provided, using a temporary file..."
Error in value[3L] :
mofapy package not found. Make sure that Reticulate is pointing to the right Python binary.
Please read the instructions here: GitHub - bioFAM/MOFA: Multi-Omics Factor Analysis
sessionInfo()
R version 3.6.3 (2020-02-29)
Platform: x86_64-conda_cos6-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /shared/mfs/data/software/miniconda/envs/r-3.6.3/lib/libopenblasp-r0.3.9.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8
[4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=en_US.UTF-8 LC_ADDRESS=en_US.UTF-8
[10] LC_TELEPHONE=en_US.UTF-8 LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=en_US.UTF-8
attached base packages:
[1] grid parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] tidygraph_1.2.0 ggraph_2.0.3 rstatix_0.5.0
[4] ggpubr_0.3.0 farver_2.0.3 devtools_2.3.0
[7] usethis_1.6.1 igraph_1.2.5 miceadds_3.9-14
[10] mice_3.9.0 colorRamps_2.3 corrr_0.4.2
[13] caret_6.0-86 lineup_0.37-11 nortest_1.0-4
[16] MOFA2_0.99.7 ggpmisc_0.3.4 org.At.tair.db_3.10.0
[19] clusterProfiler_3.14.3 gridExtra_2.3 forcats_0.5.0
[22] tibble_3.0.1 tidyverse_1.3.0 readxl_1.3.1
[25] DOSE_3.12.0 tidyr_1.1.0 purrr_0.3.4
[28] smatr_3.4-8 ggrepel_0.8.2 cowplot_1.0.0
[31] stringr_1.4.0 pathview_1.26.0 org.Hs.eg.db_3.10.0
[34] AnnotationDbi_1.48.0 gplots_3.0.3 doParallel_1.0.15
[37] iterators_1.0.12 foreach_1.5.0 kableExtra_1.1.0
[40] mixOmics_6.10.9 lattice_0.20-41 MASS_7.3-51.6
[43] corrplot_0.84 VennDiagram_1.6.20 futile.logger_1.4.3
[46] factoextra_1.0.7 FactoMineR_2.3 plyr_1.8.6
[49] readr_1.3.1 edgeR_3.28.1 limma_3.42.2
[52] DESeq2_1.26.0 xlsx_0.6.1 WGCNA_1.69
[55] fastcluster_1.1.25 dynamicTreeCut_1.63-1 dplyr_0.8.5
[58] MOFAdata_1.3.1 MultiAssayExperiment_1.12.0 SummarizedExperiment_1.16.0
[61] DelayedArray_0.12.0 BiocParallel_1.20.0 matrixStats_0.56.0
[64] Biobase_2.46.0 GenomicRanges_1.38.0 GenomeInfoDb_1.22.0
[67] IRanges_2.20.0 S4Vectors_0.24.0 BiocGenerics_0.32.0
[70] pheatmap_1.0.12 MOFA_1.2.0 data.table_1.12.8
[73] RColorBrewer_1.1-2 yaml_2.2.1 knitr_1.28
[76] ggplot2_3.3.1 reshape2_1.4.4
loaded via a namespace (and not attached):
[1] rappdirs_0.3.1 ModelMetrics_1.2.2.2 acepack_1.4.1 bit64_0.9-7
[5] rpart_4.1-15 KEGGREST_1.26.1 RCurl_1.98-1.2 generics_0.0.2
[9] preprocessCore_1.48.0 callr_3.4.3 lambda.r_1.2.4 RSQLite_2.2.0
[13] europepmc_0.3 bit_1.1-15.2 enrichplot_1.6.1 webshot_0.5.2
[17] xml2_1.3.2 lubridate_1.7.8 assertthat_0.2.1 viridis_0.5.1
[21] gower_0.2.1 xfun_0.14 hms_0.5.3 rJava_0.9-12
[25] evaluate_0.14 fansi_0.4.1 progress_1.2.2 caTools_1.18.0
[29] dbplyr_1.4.3 Rgraphviz_2.30.0 DBI_1.1.0 geneplotter_1.64.0
[33] htmlwidgets_1.5.1 rARPACK_0.11-0 ellipsis_0.3.1 RSpectra_0.16-0
[37] backports_1.1.7 annotate_1.64.0 vctrs_0.3.0 remotes_2.1.1
[41] abind_1.4-5 withr_2.2.0 ggforce_0.3.1 triebeard_0.3.0
[45] checkmate_2.0.0 prettyunits_1.1.1 cluster_2.1.0 crayon_1.3.4
[49] ellipse_0.4.1 genefilter_1.68.0 recipes_0.1.12 pkgconfig_2.0.3
[53] tweenr_1.0.1 pkgload_1.0.2 nlme_3.1-147 vipor_0.4.5
[57] nnet_7.3-14 rlang_0.4.6 lifecycle_0.2.0 modelr_0.1.8
[61] rprojroot_1.3-2 cellranger_1.1.0 polyclip_1.10-0 graph_1.64.0
[65] Matrix_1.2-18 urltools_1.7.3 carData_3.0-3 Rhdf5lib_1.8.0
[69] reprex_0.3.0 base64enc_0.1-3 beeswarm_0.2.3 processx_3.4.2
[73] ggridges_0.5.2 png_0.1-7 viridisLite_0.3.0 bitops_1.0-6
[77] pROC_1.16.2 KernSmooth_2.23-17 Biostrings_2.54.0 blob_1.2.1
[81] qvalue_2.18.0 jpeg_0.1-8.1 gridGraphics_0.5-0 ggsignif_0.6.0
[85] scales_1.1.1 leaps_3.1 memoise_1.1.0 magrittr_1.5
[89] gdata_2.18.0 zlibbioc_1.32.0 compiler_3.6.3 KEGGgraph_1.46.0
[93] cli_2.0.2 XVector_0.26.0 ps_1.3.3 htmlTable_1.13.3
[97] formatR_1.7 Formula_1.2-3 tidyselect_1.1.0 stringi_1.4.6
[101] mitools_2.4 GOSemSim_2.12.1 locfit_1.5-9.4 latticeExtra_0.6-29
[105] fastmatch_1.1-0 tools_3.6.3 rio_0.5.16 rstudioapi_0.11
[109] foreign_0.8-76 prodlim_2019.11.13 scatterplot3d_0.3-41 digest_0.6.25
[113] rvcheck_0.1.8 BiocManager_1.30.10 lava_1.6.7 Rcpp_1.0.4.6
[117] car_3.0-8 broom_0.5.6 httr_1.4.1 colorspace_1.4-1
[121] rvest_0.3.5 XML_3.99-0.3 fs_1.4.1 reticulate_1.16
[125] splines_3.6.3 graphlayouts_0.7.0 xlsxjars_0.6.1 ggplotify_0.0.5
[129] sessioninfo_1.1.1 xtable_1.8-4 jsonlite_1.6.1 futile.options_1.0.1
[133] timeDate_3043.102 corpcor_1.6.9 flashClust_1.01-2 testthat_2.3.2
[137] ipred_0.9-9 R6_2.4.1 Hmisc_4.4-0 pillar_1.4.4
[141] htmltools_0.4.0 glue_1.4.1 class_7.3-17 codetools_0.2-16
[145] fgsea_1.12.0 pkgbuild_1.0.8 curl_4.3 ggbeeswarm_0.6.0
[149] gtools_3.8.2 zip_2.0.4 openxlsx_4.1.5 GO.db_3.10.0
[153] survival_3.1-12 rmarkdown_2.1 desc_1.2.0 munsell_0.5.0
[157] DO.db_2.9 rhdf5_2.30.1 GenomeInfoDbData_1.2.2 HDF5Array_1.14.4
[161] impute_1.60.0 haven_2.2.0 gtable_0.3.0
zxue
Bonne journée
Not so easy your example script for someone who never work on R ...
I was hopping a simple script file I can run with Rscript.
But it look like the mofapy issue is solved now:
Please try Ctrl-Shit-F10 to restart rsession and test again.
Hey,
Thank you for your input. I refreshed the page, it was not the main problem. But I was able to install everything locally so I have it working now.
Thanks to everyone for helping me in these past few days.
zxue
Bonjour,
Je me permets de relancer le sujet car je suis dans la même situation décrit par zxue.
Error in value[3L] :
mofapy package not found. Make sure that Reticulate is pointing to the right Python binary.
Please read the instructions here: https://github.com/bioFAM/MOFA#installation
J'ai redémarrer ma session R et rien ne change.
Du coups es ce que quelqu'un l'utilise actuellement si oui comment fait il svp ?
Merci d'avance une bonne journée,
LK
Bonjour,
On vient de tester l'installation directement avec pip:
Ca à l'air de bien fonctionner, je viens d'essayer de charger mofapy et, pour comparer, un module qui n’existe pas dans rstudio:
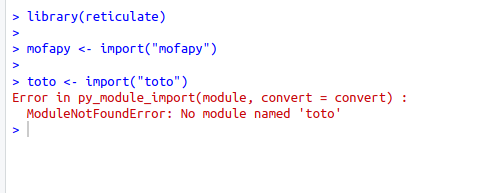
Si il y a toujours un souci, je veux bien un petit script R (a lancer avec Rscript) qui me permettrai de reproduire l'erreur