Bonjour,
j'ai une erreur de mémoire en traitant des données GCMS :
"This job was terminated because it used more memory than it was allocated."
le détail est ci-dessous :
An error occurred while running the tool toolshed.g2.bx.psu.edu/repos/yguitton/metams_rungc/metams_runGC/2.1.1.
J'utilise la version 2 de l'outil car je n'ai pas encore trouvé comment introduire les MZMl en données d'entrée sur la V3.
Il se trouve que c'est le plus gros jeu de données que j'ai jamais traité sur Galaxy, le ZIP fait 13,8Go avec beaucoup de fichiers (données GCMS basse résolution)
Comment peut-on contourner ce problème?
Merci
Guy
Bonjour Guy
navré pour le délais de réponse. Une piste pour éviter la saturation mémoire pourrait être d'utiliser un workflow un peu différent qui passe par:
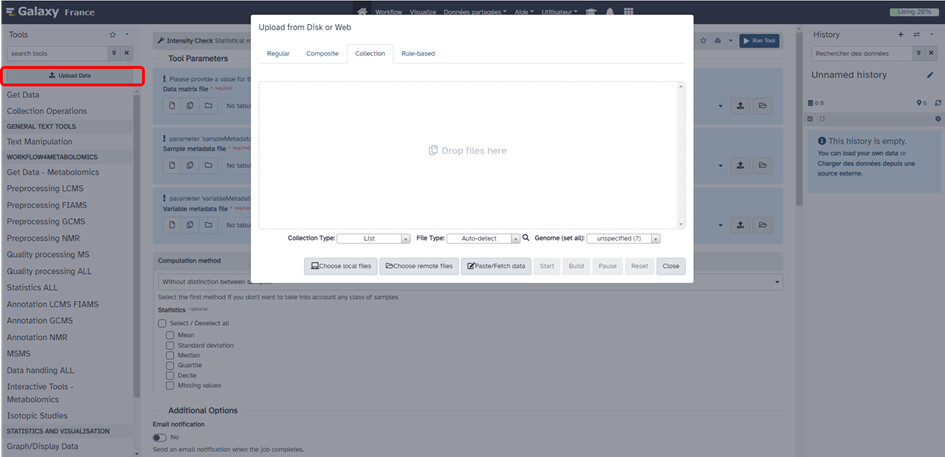
1- la création d'une data collection et non d'un zip (cf Hands-on: Hands-on: Mass spectrometry : GC-MS analysis with metaMS package / Metabolomics). Ici l'idée est de charger l'ensemble de vos fichiers mzML dans galaxy en tant que "collection" et pas regular comme vous le faisiez
2- ensuite de faire le peakpicking en utilisant XCMS (ce que fait metaMS en fait) mais ici vous le feriez étape par étape. Il y a un TP GCMS disponible sur internet ( Hands-on: Hands-on: Mass spectrometry : GC-MS analysis with metaMS package / Metabolomics) Mais ATTENTIOn il n'est pas encore finalisé et est encore plein de beugs
3- lorsque vous avez fait l'étape de peakpicking sur vos fichier et fait l'étape de merge vous avez un fichier xset.merged.RData qui peut être importé dans metaMS (les nouvelles versions) et vous pourrez ainsi reprendre le cours de vos analyses habituelles
j'espère avoir aidé
Yann