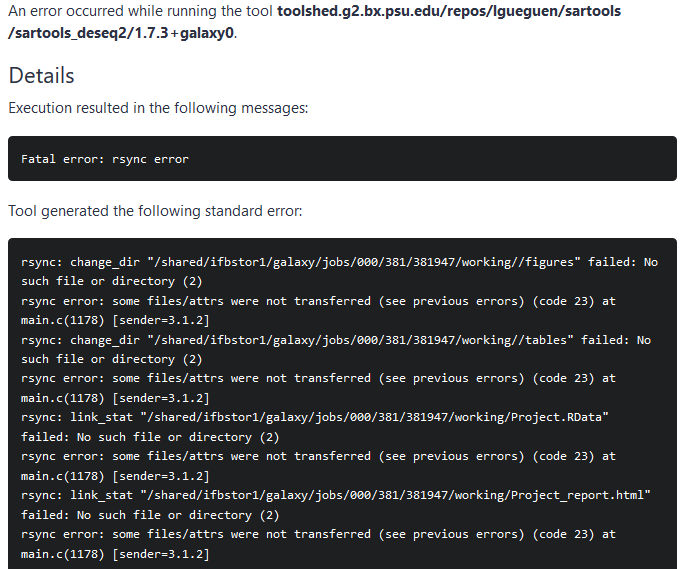
Je vous le transfère directement dans le message, car les nouveaux utilisateurs n'ont pas le droit d'uploader des fichiers :
Loading required package: DESeq2
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, basename, cbind, colnames,
dirname, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget,
order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rownames, sapply, setdiff, sort, table, tapply,
union, unique, unsplit, which.max, which.min
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:base’:
expand.grid
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: SummarizedExperiment
Loading required package: MatrixGenerics
Loading required package: matrixStats
Attaching package: ‘MatrixGenerics’
The following objects are masked from ‘package:matrixStats’:
colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
colWeightedMeans, colWeightedMedians, colWeightedSds,
colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
rowWeightedSds, rowWeightedVars
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Attaching package: ‘Biobase’
The following object is masked from ‘package:MatrixGenerics’:
rowMedians
The following objects are masked from ‘package:matrixStats’:
anyMissing, rowMedians
Loading required package: edgeR
Loading required package: limma
Attaching package: ‘limma’
The following object is masked from ‘package:DESeq2’:
plotMA
The following object is masked from ‘package:BiocGenerics’:
plotMA
Loading required package: ggplot2
Loading required package: kableExtra
Registered S3 method overwritten by 'GGally':
method from
+.gg ggplot2
----------------------------------------------
Welcome to SARTools version 1.7.3.
R template scripts are available on GitHub.
----------------------------------------------
There were 13 warnings (use warnings() to see them)
[1] "All the parameters are correct"
Target file:
label files group
Mock_M2 Mock_M2 dataset_928292.dat Mock
Mock_M3 Mock_M3 dataset_928293.dat Mock
Mock_M4 Mock_M4 dataset_928294.dat Mock
CaMV_C1 CaMV_C1 dataset_928295.dat CaMV
CaMV_C2 CaMV_C2 dataset_928296.dat CaMV
CaMV_C3 CaMV_C3 dataset_928297.dat CaMV
TuYV_T1 TuYV_T1 dataset_928298.dat TuYV
TuYV_T2 TuYV_T2 dataset_928299.dat TuYV
TuYV_T3 TuYV_T3 dataset_928300.dat TuYV
Loading files:
dataset_928292.dat: 27656 rows and 5660 null count(s)
dataset_928293.dat: 27656 rows and 5715 null count(s)
dataset_928294.dat: 27656 rows and 6050 null count(s)
dataset_928295.dat: 27656 rows and 5440 null count(s)
dataset_928296.dat: 27656 rows and 5581 null count(s)
dataset_928297.dat: 27656 rows and 5539 null count(s)
dataset_928298.dat: 27656 rows and 5818 null count(s)
dataset_928299.dat: 27656 rows and 5614 null count(s)
dataset_928300.dat: 27656 rows and 5810 null count(s)
Error in counts%%1 : non-numeric argument to binary operator
Calls: loadCountData
Execution halted