Bonjour. J'ai un problème avec l'installation d'une librairie sur Rstudio. J'essaie d'installer BiocManager::DEP mais il y aurait un soucis de dépendance ou de droit d'écriture. Je ne sais pas trop. Pouvez_vous m'aider svp ?
Voici l'erreur renvoyée par Rstudio:
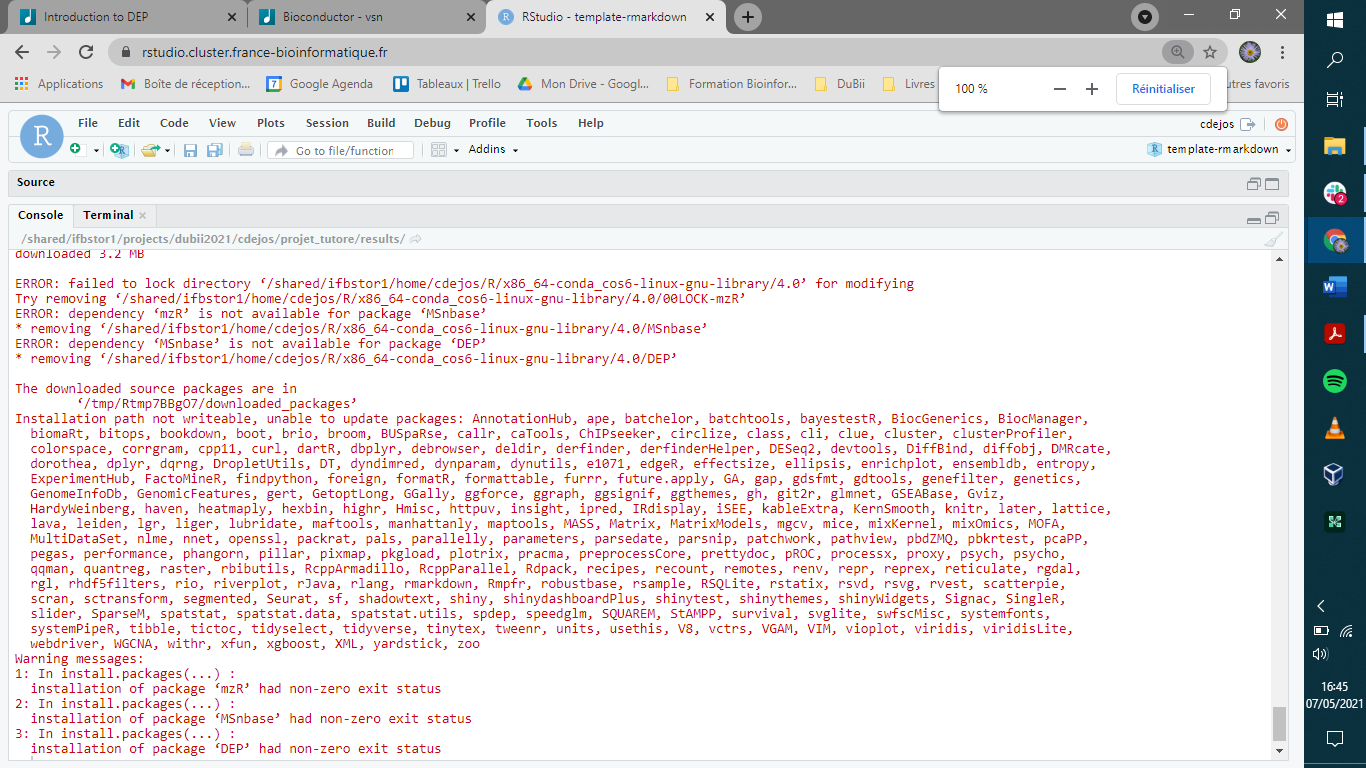
ERROR: failed to lock directory ‘/shared/ifbstor1/home/cdejos/R/x86_64-conda_cos6-linux-gnu-library/4.0’ for modifying
Try removing ‘/shared/ifbstor1/home/cdejos/R/x86_64-conda_cos6-linux-gnu-library/4.0/00LOCK-mzR’
ERROR: dependency ‘mzR’ is not available for package ‘MSnbase’
- removing ‘/shared/ifbstor1/home/cdejos/R/x86_64-conda_cos6-linux-gnu-library/4.0/MSnbase’
ERROR: dependency ‘MSnbase’ is not available for package ‘DEP’ - removing ‘/shared/ifbstor1/home/cdejos/R/x86_64-conda_cos6-linux-gnu-library/4.0/DEP’
The downloaded source packages are in
‘/tmp/Rtmp7BBgO7/downloaded_packages’
Installation path not writeable, unable to update packages: AnnotationHub, ape, batchelor, batchtools, bayestestR, BiocGenerics, BiocManager, biomaRt, bitops,
bookdown, boot, brio, broom, BUSpaRse, callr, caTools, ChIPseeker, circlize, class, cli, clue, cluster, clusterProfiler, colorspace, corrgram, cpp11, curl,
dartR, dbplyr, debrowser, deldir, derfinder, derfinderHelper, DESeq2, devtools, DiffBind, diffobj, DMRcate, dorothea, dplyr, dqrng, DropletUtils, DT,
dyndimred, dynparam, dynutils, e1071, edgeR, effectsize, ellipsis, enrichplot, ensembldb, entropy, ExperimentHub, FactoMineR, findpython, foreign, formatR,
formattable, furrr, future.apply, GA, gap, gdsfmt, gdtools, genefilter, genetics, GenomeInfoDb, GenomicFeatures, gert, GetoptLong, GGally, ggforce, ggraph,
ggsignif, ggthemes, gh, git2r, glmnet, GSEABase, Gviz, HardyWeinberg, haven, heatmaply, hexbin, highr, Hmisc, httpuv, insight, ipred, IRdisplay, iSEE,
kableExtra, KernSmooth, knitr, later, lattice, lava, leiden, lgr, liger, lubridate, maftools, manhattanly, maptools, MASS, Matrix, MatrixModels, mgcv, mice,
mixKernel, mixOmics, MOFA, MultiDataSet, nlme, nnet, openssl, packrat, pals, parallelly, parameters, parsedate, parsnip, patchwork, pathview, pbdZMQ,
pbkrtest, pcaPP, pegas, performance, phangorn, pillar, pixmap, pkgload, plotrix, pracma, preprocessCore, prettydoc, pROC, processx, proxy, psych, psycho,
qqman, quantreg, raster, rbibutils, RcppArmadillo, RcppParallel, Rdpack, recipes, recount, remotes, renv, repr, reprex, reticulate, rgdal, rgl, rhdf5filters,
rio, riverplot, rJava, rlang, rmarkdown, Rmpfr, robustbase, rsample, RSQLite, rstatix, rsvd, rsvg, rvest, scatterpie, scran, sctransform, segmented, Seurat,
sf, shadowtext, shiny, shinydashboardPlus, shinytest, shinythemes, shinyWidgets, Signac, SingleR, slider, SparseM, spatstat, spatstat.data, spatstat.utils,
spdep, speedglm, SQUAREM, StAMPP, survival, svglite, swfscMisc, systemfonts, systemPipeR, tibble, tictoc, tidyselect, tidyverse, tinytex, tweenr, units,
usethis, V8, vctrs, VGAM, VIM, vioplot, viridis, viridisLite, webdriver, WGCNA, withr, xfun, xgboost, XML, yardstick, zoo
Warning messages:
1: In install.packages(...) :
installation of package ‘mzR’ had non-zero exit status
2: In install.packages(...) :
installation of package ‘MSnbase’ had non-zero exit status
3: In install.packages(...) :
installation of package ‘DEP’ had non-zero exit status