Bonjour
Il y a peu vous avez mis à jour FROGS et il semble que, comme nous, vous avez rencontré des problèmes d'installation.
De notre côté (plateforme Migale et Genotoul), les environnements conda comprenant les dépendances phyloseq, picrust, ou deseq2, avaient des temps de création extrêmement long pour in fine ne pas se faire. Nous sommes en train d'installer manuellement ces env en utilisant une version up to date de mamba au lieu de conda.... c'est à n'y rien comprendre.
De votre côté il semble qu'aucun env ne soit créer correctement, puisque nous obtenons systématiquement des erreures command not found.
Nous avons fais une version Galaxy2 de FROGS 4.1.0, pour mettre à jour des fichiers .loc qui posaient problème (voir ensuite). Afin qu’il n’y ait pas d’interférence entre les anciens fichiers .loc et les nouveaux, il est nécessaire de désinstaller la version galaxy1, et d’installer la version galaxy2 disponible sur le toolshed.
D'autre part dans cette version nous avons renommé certains outils (.... sorry ...) et fusionné 2 outils en 1. Si vous vous souvenez, pour proposer un panel d'outils dans un ordre logique, vous aviez mis en place un tool pannel dédié
Je suppose que c’est à cause du changement de nom de ces outils que ce tool pannel bug en gardant uniquement les noms en commun entre les deux versions, et en mixant les versions. Je vous liste ci-dessous les modification (en terme de tool_id) des outils qui ont été conservés en version 4.0.1, et donc par conséquences les outils 4.1.0 qui manquent:
Tool_id version 4.0.1 Tool_id version 4.1.0
FROGS_OTU_filters FROGS_cluster_filters
FROGS_affiliation_OTU FROGS_taxonomic_affiliation
FROGS_clusters_stat FROGS_cluster_stats
FROGS_affiliations_stat FROGS_affiliation_stats
FROGSFUNC_step2_copynumbers FROGSFUNC_step3_functions
FROGSFUNC_step3_functions
FROGSFUNC_step4_pathways FROGSFUNC_step4_pathways
Nous avons également modifié les tool name (même pour les tool_id non modifié). J’ai l’impression qu’il faut qu’on vous les fournisse pour mettre à jour ce panel. Vous nous confirmez ? ex :
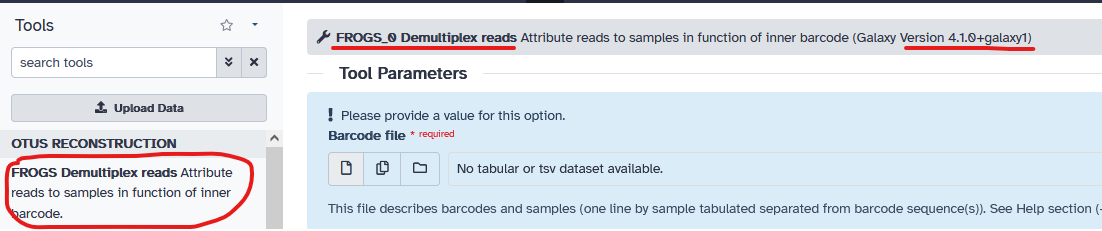
- Tool_id : FROGS_demultiplex
- Tool_name 4.0.1 : FROGS Demultiplex reads -> nom reporté dans le tool panel
- Tool_name 4.1.0 : FROGS_0 Demultiplex reads -> nom reporté dans le header de l’interface de paramétrage
Une solution (mais c’est vous les experts), serait peut-être de faire 2 tool panels en fonction de la version ? ou bien ne conserver que la version 4.1.0. Au cas où cette solution serait acceptable je vous mets ci-dessous les tool_id 4.1.0 dans l’ordre le plus pertinent (voir aussi section Simplest way / integrated_tool_panel.xml dans GitHub - geraldinepascal/FROGS-wrappers: Galaxy wrappers for FROGS) :
- FROGS_demultiplex
- FROGS_preprocess
- FROGS_clustering
- FROGS_remove_chimera
- FROGS_cluster_filters
- FROGS_itsx
- FROGS_taxonomic_affiliation
- FROGS_affiliation_filters
- FROGS_affiliation_postprocess
- FROGS_normalisation
- FROGS_Tree
- FROGS_cluster_stats
- FROGS_affiliation_stats
- FROGS_biom_to_stdBiom
- FROGS_biom_to_tsv
- FROGS_tsv_to_biom
- FROGSSTAT_Phyloseq_Import_Data
- FROGSSTAT_Phyloseq_Composition_Visualisation
- FROGSSTAT_Phyloseq_Alpha_Diversity
- FROGSSTAT_Phyloseq_Beta_Diversity
- FROGSSTAT_Phyloseq_Sample_Clustering
- FROGSSTAT_Phyloseq_Structure_Visualisation
- FROGSSTAT_Phyloseq_Multivariate_Analysis_Of_Variance
- FROGSSTAT_DESeq2_Preprocess
- FROGSSTAT_DESeq2_Visualisation
- FROGSFUNC_step1_placeseqs
- FROGSFUNC_step3_functions
- FROGSFUNC_step4_pathways
Pour finir, petite note sur la gestion des fichiers .loc qui posaient problème.
Il y a 3 fichiers .loc qui remplacent 3 anciens, pour les outils FROGSFUNC. Ce sont les mêmes que les anciens avec juste une nouvelle colonne en plus qui indique la version de picrust2 actuelle.
Les noms des fichiers sont devenus les suivants :
frogs_picrust2_default_dir.loc -> frogs_picrust2_placeseqs.loc
frogs_picrust2_marker_table.loc -> frogs_picrust2_functions.loc
frogs_picrust2_pathway_map.loc -> frogs_picrust2_pathways.loc
Le fichier de configuration config/shed_tool_data_table_conf.xml décrit l’emplacement de ces fichiers .loc.
Dans ces fichiers, il suffit de décommenter la partie inférieure et de remplacer les variables d’environnement <Galaxy_dir> et <PICRUSt2_env> pour avoir le bon emplacement des fichiers.
Exemple avec le fichier frogs_picrust2_placeseqs.loc des parties à remplacer :
#picrust2_default_dir_16S 16S <Galaxy_dir>/database/dependencies/_conda/envs/<PICRUSt2_env>/lib/python3.6/site-packages/picrust2/default_files/prokaryotic/pro_ref/ <Galaxy_dir>/database/dependencies/_conda/envs/<PICRUSt2_env>/lib/python3.6/site-packages/picrust2/default_files/16S_counts.txt.gz epa-ng 2.5.1
#picrust2_default_dir_16S 16S <Galaxy_dir>/database/dependencies/_conda/envs/<PICRUSt2_env>/lib/python3.6/site-packages/picrust2/default_files/prokaryotic/pro_ref/ <Galaxy_dir>/database/dependencies/_conda/envs/<PICRUSt2_env>/lib/python3.6/site-packages/picrust2/default_files/16S_counts.txt.gz sepp 2.5.1
#picrust2_default_dir_ITS ITS <Galaxy_dir>/database/dependencies/_conda/envs/<PICRUSt2_env>/lib/python3.6/site-packages/picrust2/default_files/fungi/fungi_ITS/ <Galaxy_dir>/database/dependencies/_conda/envs/<PICRUSt2_env>/lib/python3.6/site-packages/picrust2/default_files/fungi/ITS_counts.txt.gz epa-ng 2.5.1
#picrust2_default_dir_18S 18S <Galaxy_dir>/database/dependencies/_conda/envs/<PICRUSt2_env>/lib/python3.6/site-packages/picrust2/default_files/fungi/fungi_18S/ <Galaxy_dir>/database/dependencies/_conda/envs/<PICRUSt2_env>/lib/python3.6/site-packages/picrust2/default_files/fungi/18S_counts.txt.gz epa-ng 2.5.1
Je suis navré de cette situation, tous les planemo tests fonctionnent parfaitement et le github action qui suit les reco de l’IUC aussi. Nous ne comprenons pas pourquoi nous avons autant de difficultés à installer la suite d’outils sur nos serveurs.
Nous pouvons en reparler de vives voix si cela est préférable.
@mariabernard, @vindarbot (adresse de contact frogs-support@inrae.fr )