Hello dear Cluster Team,
I d like your help to trace the problem of my last series of jobs, that Failed but without any specific error message in the log files of the tool "STAR", or of "nextflow".
It's about
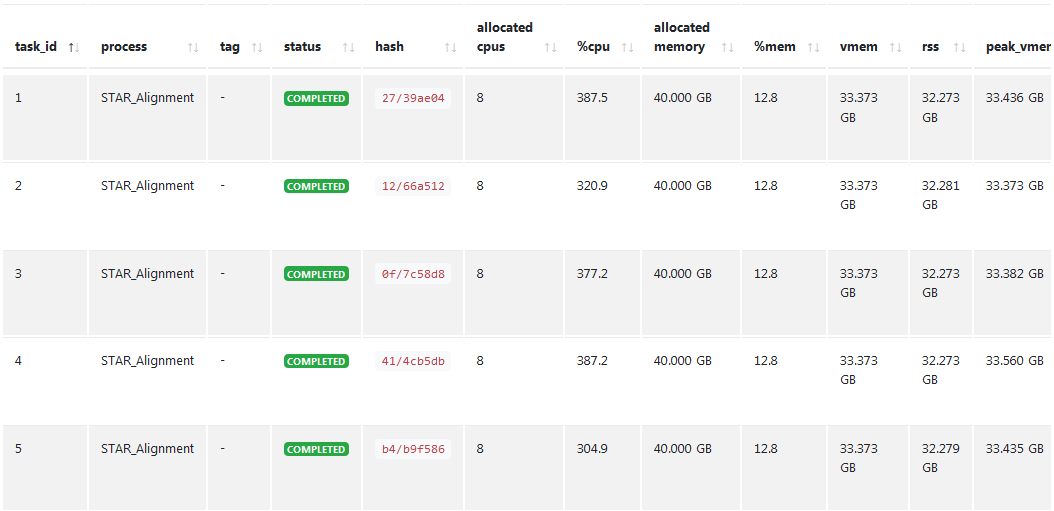
jobIDs = { 13466845 - 51}, -u mkondili, on 17.October.
My doubts are mainly on the memory demanded and used from tool.
The tool STAR, which I use for alignment of fastq-files, requires 40Gb of memory, which I declare in the process when I call it.
But then, in the Sbatch I declare the following :
#SBATCH -A mbnl_dct
#SBATCH --mem 40GB
#SBATCH -n 1
#SBATCH -c 8
on the contrary, for nextflow to launch and schedule the jobs in the cores, I need max 10Gb,
but when I tried with
#SBATCH --mem 10GB the jobs failed anyway.
So I have some questions too.
Is there a limit by default for very "demanding" tools, in the cluster, for which my script exceeds ?
Or an error in the way I run the sbatch /nextflow ?
I am not sure how many cores to ask for 40Gb. Should there be an equivalence of
cores x mem/core = total Sbatch --mem ?
Is the Sbatch --mem parameter referring to the memory used by nextflow ,or the total memory by all the processes happening in main pipeline ?
I should also learn how to calculate the memory each tool consumes, to optimise my scripts.
If you have any tools/commands in the cluster for that, please share.
Thanks in advance,
I hope I have explained enough to help you understand.
Let me know if you have further questions.